In pharma chemical development one deals with all sort of molecules. In the company where I was gainfully employed research often produced lots of heterocyclic compounds, usually containing lots of nitrogen. Looking at the structures they were all flat and aromatic and insoluble, probably they would have made better explosives than pharmaceuticals. So imagine my surprise when a molecule with some 3D characteristics and only containing 1 nitrogen landed on my desk. This was some molecule with lots of chiral centers (13 to be exact) also present were 4 C=C bonds, 3 of them cis and one of them tri-substituted! So we won’t discuss the potential of isomers as there are lots of them. My job was to come up with 1) a scalable, reproducible process and 2) a novel synthetic route.
One of the many steps was the boron mediated aldol reaction shown below:
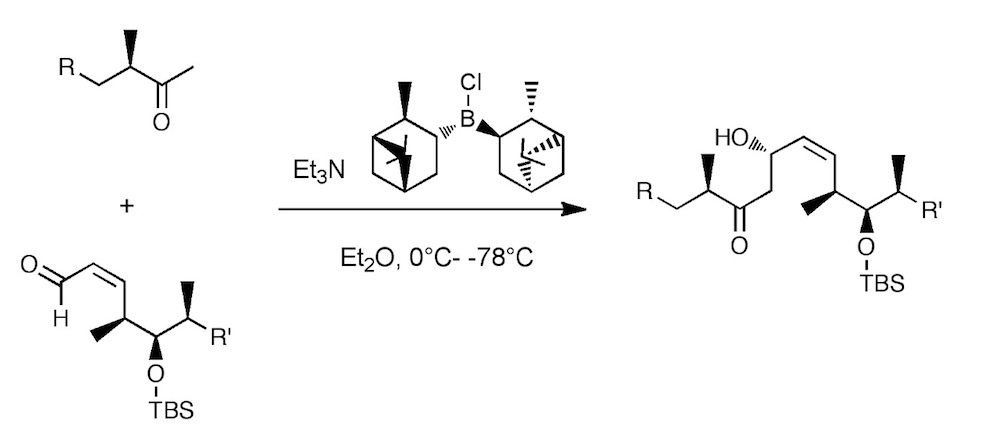
Now this turned out to be one of the most difficult reactions I have ever attempted to scale-up! This aldol reaction uses reagent control to overcome the natural stereochemistry, that is, it is a mis-matched reaction. Further, the procedure called for a ten-fold excess of the ketone enolate, this together with the lowish yield made the desired product one of the side products. The yield of this reaction was also a problem in that is varied from 30% to 60%, when the reaction actually worked. One of the reasons for this was the quality of the DIP-Cl.
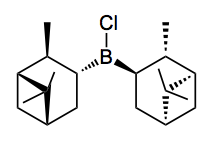 DIP-Cl: chlorobis((1R,2S,3R,5R)-2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)borane.
DIP-Cl: chlorobis((1R,2S,3R,5R)-2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)borane.
I initially used commercially available solid (+)-DIP-Cl. This reagent is difficult to obtain and to handle in large quantities, as it is hygroscopic and inherently unstable. On storage it eliminates pinene, which reduces the quality of the reagent and produces undefined boron species. Obtaining a well-defined quality reagent on a large scale from a commercial supplier was problematic. Routine analytical methods are not really suitable for monitoring the quality of this boron reagent and the only method of testing its quality it to use it! When the reaction worked with this solid DIP-Cl I did not obtain the desired product but the compound containing the trans double bond together with its epimer in a 3:1 ratio.
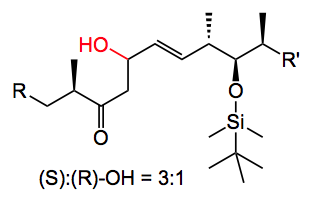
The mechanism of this isomerisation is not clear, so I am open to suggestions. There were also a few other side products, the trans allylic alcohol for example, but they were minor. I finally found a supplier that delivered this compound as a 70% solution in hexane. It came in a nice steel cylinder with a three way valve on the top which allowed the easy withdrawal of the compound while keeping it under argon. So by placing it on a balance you could weigh out exact amounts of the stuff. Anyway carrying out the aldol reaction with this material solved the reproducibility problems, the reaction worked and I actually obtained the correct product and its epimer with no C=C isomerisation. The yield was 50%.
The time had now come to attempt the first large scale reaction, 50 g of aldehyde. Utilising the conditions I had worked out on the small scale gave 23% yield of the aldol! What happened? I actually obtained significant reduction of the aldehyde back to the allylic alcohol which could not be re-isolated from the vast excesses of the reagent. It was surmised that the reason for this result was incomplete enolisation of ketone (2 hours 0°C) causing the (+)-DIP-Cl to simply reduce the aldehyde. I examined the enolisation time on a small scale and extended it to 16 hours at 0°C. Once again on the 50g scale 23% yield of aldol was obtained, this time with no reduction observed. So something else was going on, back to the drawing board. Here I was, in great danger of not being able to produce the required quantities of drug substance ordered for the clinical trials. I examined the fate of the aldol product at every stage of the process. Before work-up the desired product is formed in 65% yield together with the epimer in 33% yield. That is the aldol reaction proceeds almost quantitatively! The reaction solvent is diethyl ether (no other solvent produced a viable result) therefore safety considerations demand that before the standard oxidative work-up the solvent must be changed (ether peroxides). After an aqueous quench, solvent evaporation and re-dissolution in methylene chloride examination of the fate of the product revealed that some 20% less was present in the mixture. Carrying out the oxidative work-up resulted in a further 15 – 20% loss. The product was not stable (in the reaction mixture) to the work-up conditions. To overcome this problem I simply omitted these steps and after quenching the reaction mixture with water it was diluted with the chromatography solvent eluent and chromatographed directly on reverse-phase silica-gel. This led to a 60% yield of the desired alcohol epimer free! The epimer was isolated by further elution and re-cycled. The summarised column conditions are outlined in the following table.
Chromatography conditions.
|
700g reaction mixture |
dilute with 368kg acetonitrile/t-butylmethyl ether/water 85/15/10 |
|
apply to |
120x30cm column packed with 20kg RP-18 silica-gel |
|
elute |
with 1060kg acetonitrile/t-butylmethyl ether/water 85/15/10 |
|
change eluent to |
acetonitrile/t-butylmethyl ether 1/1 |
|
fractionate |
20kg fractions (8) |
Combining and evaporating product fractions to 10% of original volume followed by extraction with ethyl acetate and re-evaporation to dryness provides 150g of the pure aldol product. The purification procedure is not optimal. For the production campaign I required some 20m3 of solvent.
So the moral of this story is keep trying. My all your reagents be pure and stable.
17,088 total views, 1 views today